Biosimilars can expand patient access to important cost‑effective therapies
While effective and necessary, biologics can be costly
Biologics account for >40% of all US prescription drug spending but account for only a small percentage of prescription drug use1,2
In 2021 alone, US biologic
spending totaled
$256 billion3
In 2021, Medicare Part B spent over $4 billion on intravitreal anti-VEGF therapy4*
OVER $1 BILLION
Ranibizumab
OVER $3 BILLION
Aflibercept
Ranibizumab and aflibercept accounted for 11% of Medicare Part B spend before the entry of biosimilars.4
*Expenditure cites Medicare Part B only. It does not include spend by other government and commercial payers, such as spend for Medicare Advantage members.
The Biologics Price Competition and Innovation (BPCI) Act of 2009 was enacted as part of the Affordable Care Act, which created an FDA registration pathway for biologics to demonstrate biosimilarity, as a concerted effort to5,6:

Provide more
treatment options

Lower healthcare costs
through competition

Increase access to
lifesaving medications
The rigorous FDA biosimilars approval pathway
The BPCI Act established a rigorous registration pathway for biosimilars in the US, promoting the
development of more cost-effective treatment options, without compromising efficacy and safety.5,6

Reference product development
Demonstrate safety and effectiveness with adequate and well-controlled substantial evidence for a new product.6
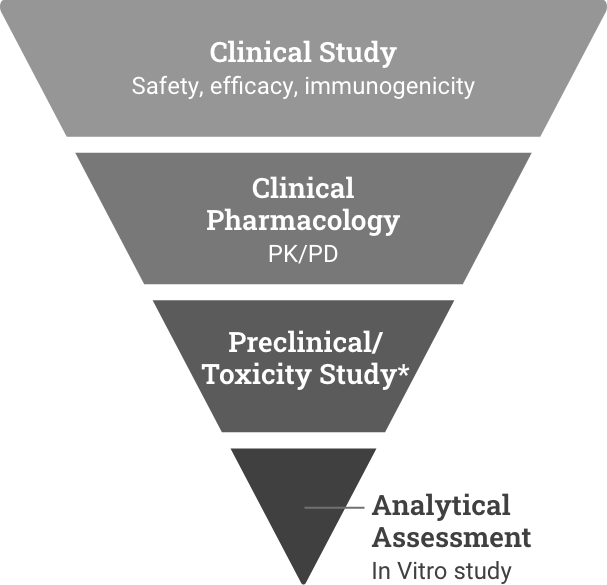
Biosimilar development
Demonstrate high similarity to reference product with no clinically meaningful difference in safety, purity, and potency via in vitro, in vivo, and clinical studies.7

*Preclinical refers to the reference product approval pathway. Toxicity study refers to the biosimilar approval pathway and could be conducted as part of the analytical or clinical study.
Biosimilars are tracked as part of a post-market surveillance
to ensure continued safety beyond manufacturing.8
Biosimilars increase choice in the marketplace, resulting in realized and projected savings for the healthcare system
The first biosimilar was approved in 2015. As of July 2023, at least 40 biosimilars have been approved by the FDA across a wide range of therapeutic areas, including9:
- Oncology
- Endocrinology
- Rheumatology
For ophthalmology, CIMERLI® is the first and only FDA-approved biosimiliar interchangeable with Lucentis® (ranibizumab injection) for all indications.11
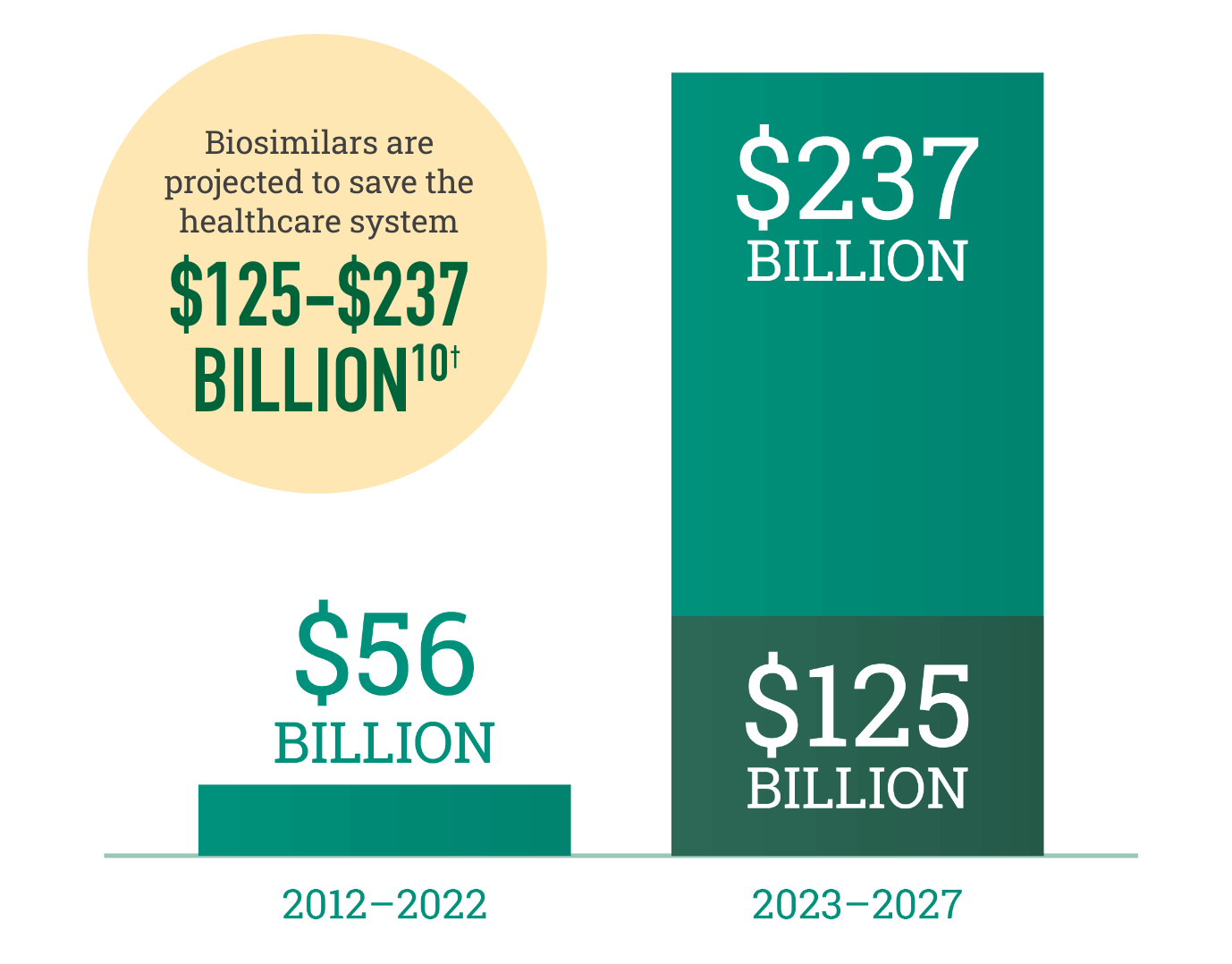
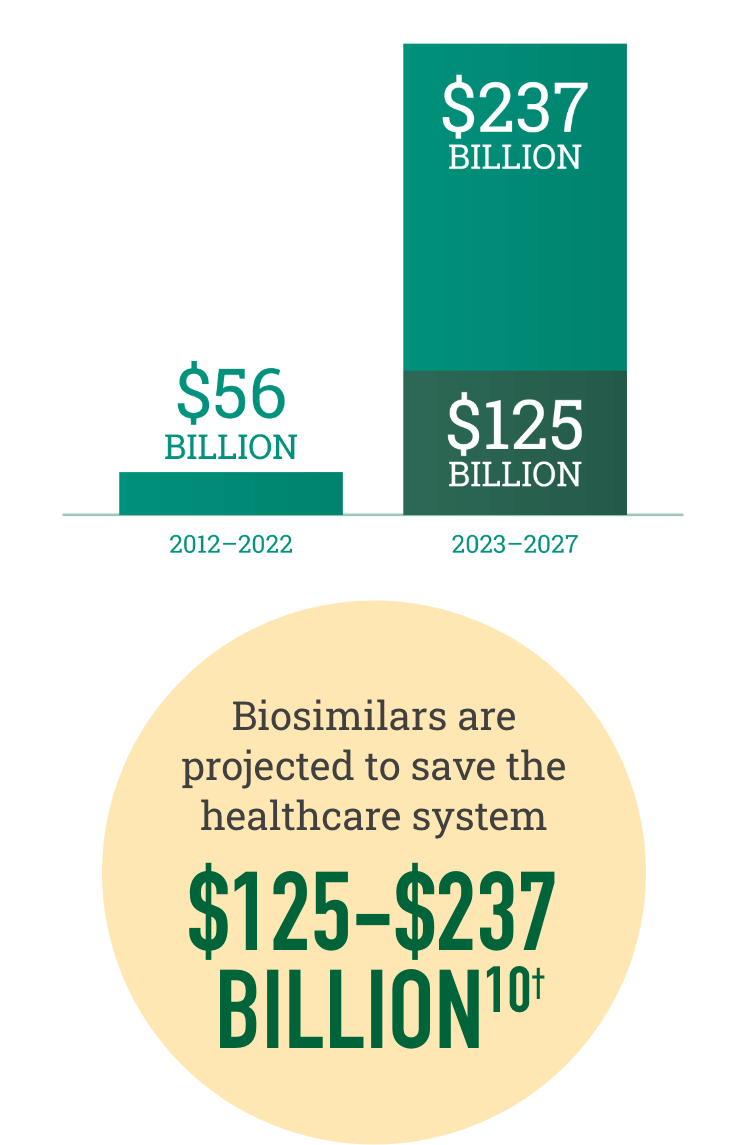
†Savings over the next 5 years are dependent on government policy changes to support adoption.
CIMERLI® was proven clinically equivalent to Lucentis® (ranibizumab injection) in terms of efficacy and safety12
See Head-to-head DataSandoz One Source provides comprehensive practice and patient support
Discover Sandoz One Source SupportRequest a visit from a Sandoz representative
Connect With Us